


Recent Findings and Current Research for Shwachman-Diamond Syndrome
What is Shwachman-Diamond Syndrome?
Shwachman-Diamond Syndrome (SDS) is an autosomal dominant and recessive multisystem disorder. Depending on the gene, an individual needs to possess both mutated copies or just one copy to express the condition.1 SDS is primarily characterized by bone marrow failure, exocrine pancreatic insufficiency (EPI) and skeletal abnormalities (Figure 1). These features translate into symptoms of poor feeding, slow growth, and intellectual disability. Furthermore, SDS patients often present with cytopenia, a reduction in the number of mature blood cells. The most common type of cytopenia is neutropenia, a deficiency in the number of neutrophils, which is the most abundant type of white blood cell that phagocytose pathogens. This is why SDS patients have an increased risk of contracting infections. SDS patients are also at high risk for developing myelodysplastic syndrome and acute myeloid leukemia, which are two types of blood cancers.2 SDS is the third most common inherited bone marrow failure syndrome, with a global incidence rate of approximately 1 in 77,000 births. Additionally, it is the second most common cause of EPI following cystic fibrosis.1,2 Given the severity of its symptoms and the considerable impact they can have on an individuals’ quality of life, coupled with the lack of cure and the average lifespan from diagnosis is 35 years, further research needs to be conducted to learn about the genetic causes. Doing so will allow for increased accuracy in diagnostic methods and will direct the development of more effective treatments or a cure.
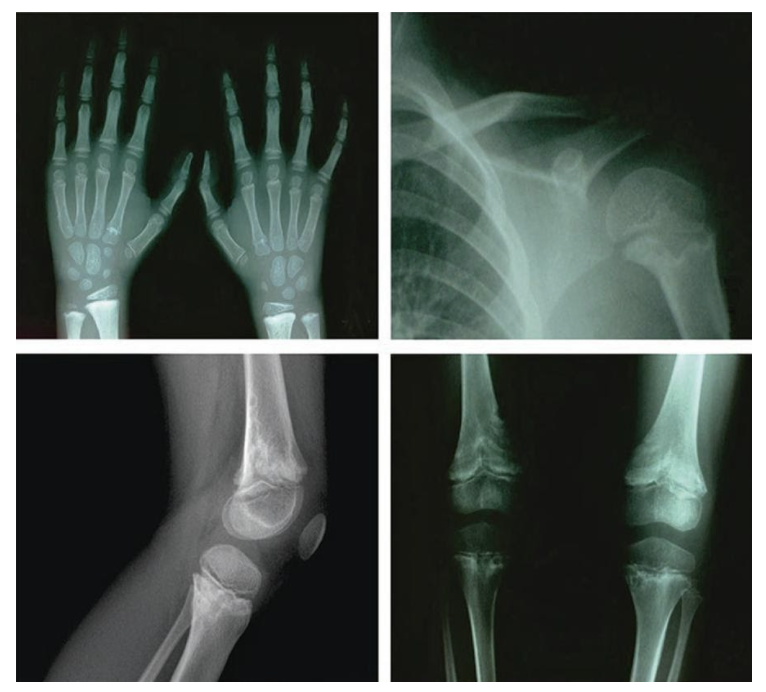
Figure 1: X-ray of skeletal and joint abnormalities in a SDS patient.
Genetic Basis
Currently, about 90% of SDS cases can be linked to mutations in 1 of 4 genes. The other 10% remains unaccounted for. Of the four genes, Shwachman-Bodian-Diamond Syndrome (SBDS) gene on chromosome 7 is the most recognized (Figure 2). SBDS is a highly conserved protein across archaea and eukaryotes and is highly expressed in mammalian tissues. To be diagnosed with SDS, an individual must be homozygous recessive for the mutated SBDS alleles.4 SBDS mutations usually result in reduced protein levels through impaired ribosome biogenesis. SBDS mutations have been linked to impaired associations between the 40S and 60S ribosomal subunits that assemble in the cell cytoplasm to produce proteins.5 Additionally, studies have suggested a role of SBDS in reactive oxygen species regulation, the cellular stress response, and hematopoiesis (the process of forming blood cells).

Figure 2: Location of SBDS gene on chromosome 7.
Past Research and Knowledge
Past research regarding SDS has mainly focused on two areas. The first has been understanding and summarizing the clinical presentations and risks of patients diagnosed with or suspected to have SDS. These include short stature and increased risk for certain blood cancers. Currently, the only existing treatments are those aimed at managing symptoms, as there is no definitive cure to eradicate the disease. These treatments include the replacement of missing pancreatic digestive enzymes alongside a special diet of fat-soluble vitamins, blood transfusions, and antibiotics. In severe cases, hematopoietic stem cell transfusions have been used, but this is not a long-term solution.
1
The second research focus has been determining the genetic cause of SDS due to the suspected genetic inheritance pattern. It was only in 2003 that a mutation in the SBDS gene was finally identified to cause SDS. Since then, it has accounted for 80-90% of SDS cases, although there were concerns over the remaining 10-20% of patients who showed normal functioning SBDS genes.
4
Subsequently, this led to research aimed at finding other potential candidate genes, which has yielded success as discussed in the following section.
Current Research and New Findings
Current research aims to meet two goals. The first is to understand the transcriptional consequences of SBDS gene deficiency during hematopoietic differentiation. This can be investigated by examining the binding targets of the SBDS gene to gain an understanding of the mechanism by which SBDS regulates hematopoiesis. This requires additional research before a definitive conclusion can be made. However, several researchers have proposed that SBDS deficiency affects hematopoietic development at the induced pluripotent stem cell (iPSC) and early hematopoietic progenitor stages through the dysregulation of the downstream and numerous pathways that comprise these stages.
6
The second goal is to identify the other genes responsible for SDS that account for the 10-20% of cases not linked to SBDS deficiency. The status of research in this area has come further as DNAJC21, EFL1, and SRP54 have been recently identified as the genetic causes of SDS in a small proportion of cases. Like the SBDS gene, two mutated alleles of the DNAJC21 and EFL1 genes must be inherited from both parents for an individual to have SDS.
7,8
The SRP54 gene shows a dominant inheritance pattern, which means the individual only needs to inherit the mutated gene from one affected parent or, more commonly, the mutations can occur de novo without a family history of the disease.
9
However, there is still a small proportion of cases who exhibit clinical presentation of SDS without exhibiting these known mutations, which suggests that there may be more genes responsible for SDS, yet to be identified.
Conclusion
The most promising consequence of identifying all the genes responsible for SDS is that gene therapies can be developed to correct/replace the target gene in affected individuals, thus allowing them to live longer, symptom-free lives. Identifying these genes also has the potential to allow for earlier detection via amniocentesis and chorionic villus sampling, resulting in better preparedness before the birth of the affected child. Furthermore, understanding the pathways in hematopoiesis that are impacted by SBDS deficiencies can facilitate the development of other therapies to correct these abnormalities. More extensive research must be done to determine the exact pathways impacted. The research which aims to study the pathways of hematopoiesis impacted by SBDS deficiencies will also enhance the current knowledge of the role of iPSCs in embryonic development, particularly through early hematopoietic development. This ultimately shows the potential that iPSCs can have beyond their typical application for therapeutic purposes. They have become an integral part of current and, possibly, future research not only in SDS but also in investigating other genetic disorders.
Daniel D'Souza
References
- “Shwachman-Diamond syndrome,” Genetic and Rare Diseases Information Center. [Online]. Available: https://rarediseases.info.nih.gov/diseases/4863/shwachman-diamond-syndrome.
- Dror Y. Shwachman-Diamond syndrome. Pediatr Blood Cancer. 2005;45(7):892-901. doi:10.1002/pbc.20478
- Alves C, Fernandes JC, Sampaio S, et al. Shwachman-Diamond syndrome: first molecular diagnosis in a Brazilian child. Revista Brasileira de Hematologia e Hemoterapia. 2013;35(4):290-292. doi: 10.5581/1516-8484.20130058
- Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33(1):97-101. doi:10.1038/ng1062
- Ganapathi KA, Austin KM, Lee CS, et al. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007;110(5):1458-1465. doi:10.1182/blood-2007-02-075184
- Tulpule A, Kelley JM, Lensch MW, et al. Pluripotent stem cell models of Shwachman-Diamond syndrome reveal a common mechanism for pancreatic and hematopoietic dysfunction. Cell Stem Cell. 2013;12(6):727-736. doi:10.1016/j.stem.2013.04.002
- Dhanraj S, Matveev A, Li H, et al. Biallelic mutations in DNAJC21 cause Shwachman-Diamond syndrome. Blood. 2017;129(11):1557-1562. doi:10.1182/blood-2016-08-735431
- Tan S, Kermasson L, Hoslin A, et al. EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood. 2019;134(3):277-290. doi:10.1182/blood.2018893404
- Bellanné-Chantelot C, Schmaltz-Panneau B, Marty C, et al. Mutations in the SRP54 gene cause severe congenital neutropenia as well as Shwachman-Diamond-like syndrome. Blood. 2018;132(12):1318-1331. doi:10.1182/blood-2017-12-820308
Cite This Article:
D'Souza D., Speagle M., Patel M. Recent Findings and Current Research for Shwachman-Diamond Syndrome. Illustrated by C. Qian. Rare Disease Review. April 2021.
DOI: 10.13140/RG.2.2.22346.29125



